
Target:Ataxin-1
Fields:Notch signaling pathway;Spinocerebellar ataxia;Pathways of neurodegeneration - multiple diseases
Gene Name:ATXN1
Protein Name:Ataxin-1
Human Gene Id:6310
Human Swiss Prot No:P54253
Mouse Gene Id:20238
Mouse Swiss Prot No:P54254
Immunogen:The antiserum was produced against synthesized peptide derived from human Ataxin 1 around the phosphorylation site of Ser776. AA range:742-791
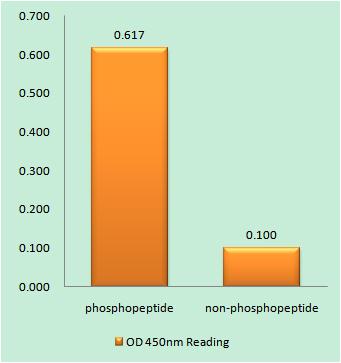
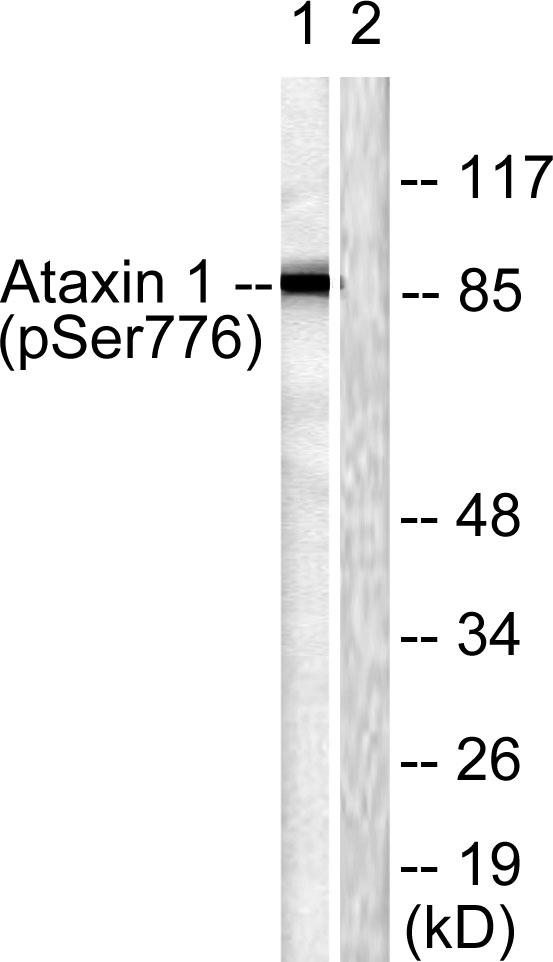
Specificity:Phospho-Ataxin-1 (S776) Polyclonal Antibody detects endogenous levels of Ataxin-1 protein only when phosphorylated at S776.
Formulation:Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Source:Polyclonal, Rabbit,IgG
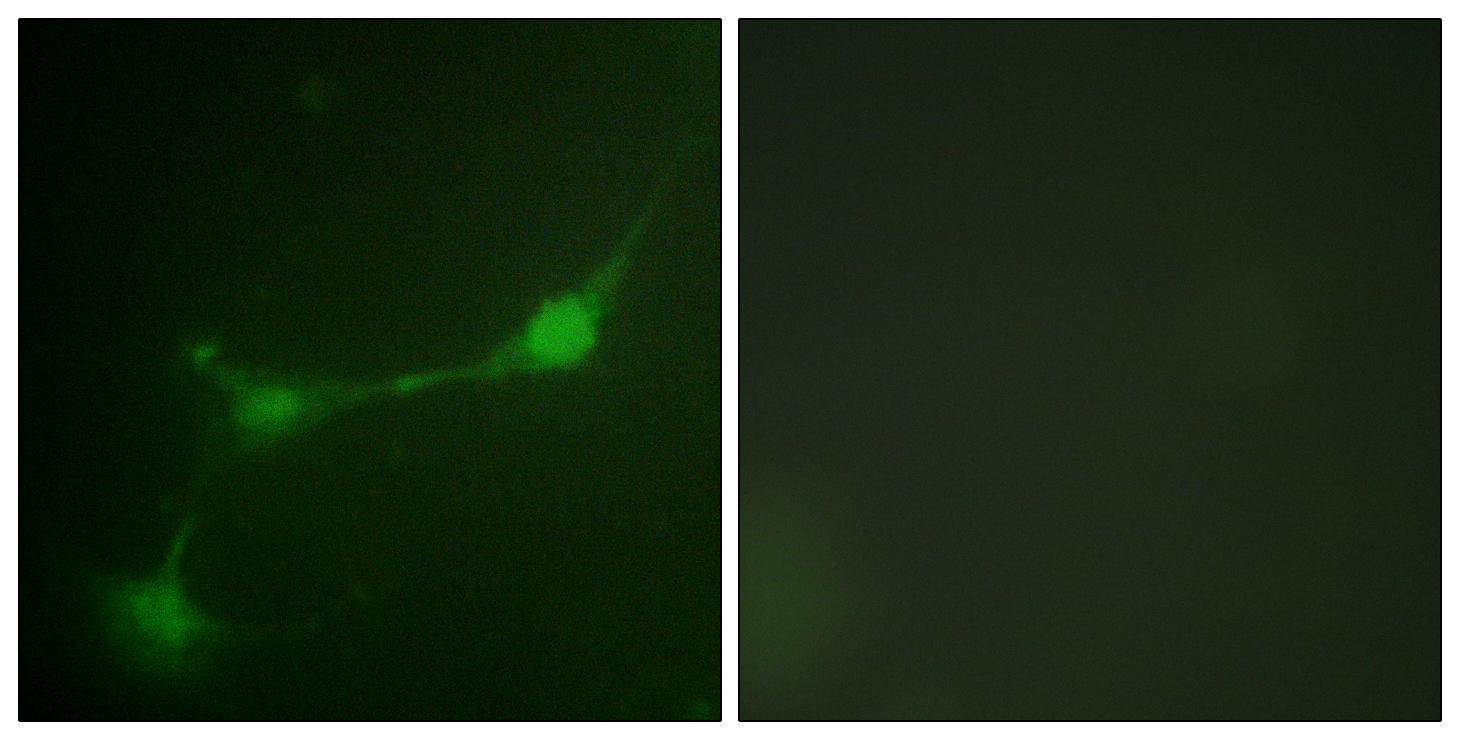
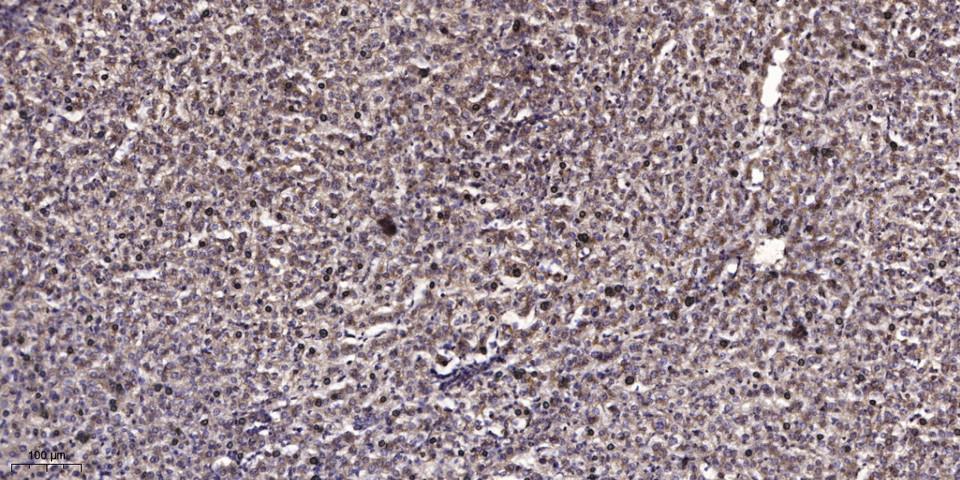
Dilution:WB 1:500 - 1:2000. IHC 1:100 - 1:300. IF 1:200 - 1:1000. ELISA: 1:10000. Not yet tested in other applications.
Purification:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Concentration:1 mg/ml
Storage Stability:-15°C to -25°C/1 year(Do not lower than -25°C)
Other Name:ATXN1;ATX1;SCA1;Ataxin-1;Spinocerebellar ataxia type 1 protein
Observed Band(KD):87kD
Background:ataxin 1(ATXN1) Homo sapiens The autosomal dominant cerebellar ataxias (ADCA) are a heterogeneous group of neurodegenerative disorders characterized by progressive degeneration of the cerebellum, brain stem and spinal cord. Clinically, ADCA has been divided into three groups: ADCA types I-III. ADCAI is genetically heterogeneous, with five genetic loci, designated spinocerebellar ataxia (SCA) 1, 2, 3, 4 and 6, being assigned to five different chromosomes. ADCAII, which always presents with retinal degeneration (SCA7), and ADCAIII often referred to as the `pure' cerebellar syndrome (SCA5), are most likely homogeneous disorders. Several SCA genes have been cloned and shown to contain CAG repeats in their coding regions. ADCA is caused by the expansion of the CAG repeats, producing an elongated polyglutamine tract in the corresponding protein. The expanded repeats are variable in size and unstable, usually increasing in size when transmitted
Function:alternative products:At least 2 isoforms are produced,disease:Defects in ATXN1 are the cause of spinocerebellar ataxia type 1 (SCA1) [MIM:164400]; also known as olivopontocerebellar atrophy I (OPCA I or OPCA1). Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to cerebellum degeneration with variable involvement of the brainstem and spinal cord. SCA1 belongs to the autosomal dominant cerebellar ataxias type I (ADCA I) which are characterized by cerebellar ataxia in combination with additional clinical features like optic atrophy, ophthalmoplegia, bulbar and extrapyramidal signs, peripheral neuropathy and dementia. SCA1 is caused by expansion of a CAG repeat in the coding region of ATXN1. Longer expansions result in earlier
Subcellular Location:Cytoplasm . Nucleus . Colocalizes with USP7 in the nucleus. .
Expression:Widely expressed throughout the body.
商品信息已成功复制,启研竭诚为您服务




