
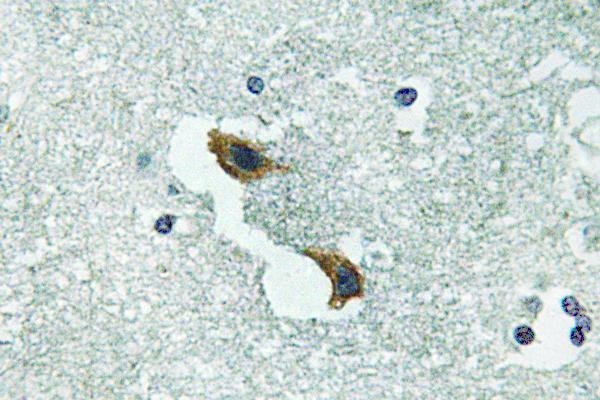

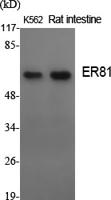
Target:ATP7A
Fields:Platinum drug resistance;Mineral absorption
Gene Name:ATP7A
Protein Name:Copper-transporting ATPase 1
Human Gene Id:538
Human Swiss Prot No:Q04656
Mouse Gene Id:11977
Mouse Swiss Prot No:Q64430
Rat Gene Id:24941
Rat Swiss Prot No:P70705
Immunogen:The antiserum was produced against synthesized peptide derived from human ATP7A. AA range:591-640
Specificity:ATP7A Polyclonal Antibody detects endogenous levels of ATP7A protein.
Formulation:Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Source:Polyclonal, Rabbit,IgG
Dilution:IHC 1:100 - 1:300. ELISA: 1:40000.. IF 1:50-200
Purification:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Concentration:1 mg/ml
Storage Stability:-15°C to -25°C/1 year(Do not lower than -25°C)
Other Name:ATP7A;MC1;MNK;Copper-transporting ATPase 1;Copper pump 1;Menkes disease-associated protein
Molecular Weight(Da):170kD
Background:ATPase copper transporting alpha(ATP7A) Homo sapiens This gene encodes a transmembrane protein that functions in copper transport across membranes. This protein is localized to the trans Golgi network, where it is predicted to supply copper to copper-dependent enzymes in the secretory pathway. It relocalizes to the plasma membrane under conditions of elevated extracellular copper, and functions in the efflux of copper from cells. Mutations in this gene are associated with Menkes disease, X-linked distal spinal muscular atrophy, and occipital horn syndrome. Alternatively-spliced transcript variants have been observed. [provided by RefSeq, Aug 2013],
Function:catalytic activity:ATP + H(2)O + Cu(2+)(In) = ADP + phosphate + Cu(2+)(Out).,disease:Defects in ATP7A are the cause of Menkes disease (MNKD) [MIM:309400]; also known as kinky hair disease. MNKD is an X-linked recessive disorder of copper metabolism characterized by generalized copper deficiency. MNKD results in progressive neurodegeneration and connective-tissue disturbances: focal cerebral and cerebellar degeneration, early growth retardation, peculiar hair, hypopigmentation, cutis laxa, vascular complications and death in early childhood. The clinical features result from the dysfunction of several copper-dependent enzymes.,disease:Defects in ATP7A are the cause of occipital horn syndrome (OHS) [MIM:304150]; also known as X-linked cutis laxa. OHS is an X-linked recessive disorder of copper metabolism. Common features are unusual facial appearance, skeletal abnormalities, chronic diarrh
Subcellular Location:Golgi apparatus, trans-Golgi network membrane ; Multi-pass membrane protein . Cell membrane ; Multi-pass membrane protein . Melanosome membrane ; Multi-pass membrane protein . Early endosome membrane ; Multi-pass membrane protein . Cell projection, axon . Cell projection, dendrite . Cell junction, synapse, postsynaptic density . Cycles constitutively between the TGN and the plasma membrane (PubMed:9147644). Predominantly found in the TGN and relocalized to the plasma membrane in response to elevated copper levels. Targeting into melanosomes is regulated by BLOC-1 complex (By similarity). In response to glutamate, translocates to neuron processes with a minor fraction at extrasynaptic sites (By similarity). .; [Isoform 3]: Cytoplasm, cytosol .; [Isoform 5]: Endoplasmic reticulum .
Expression:Widely expressed including in heart, brain, lung, muscle, kidney, pancreas, and to a lesser extent placenta (PubMed:8490646, PubMed:8490659). Expressed in fibroblasts, aortic smooth muscle cells, aortic endothelial cells and umbilical vein endothelial cells (at protein level) (PubMed:16371425). ; [Isoform 3]: Expressed in cerebellum and brain cortex.
商品信息已成功复制,启研竭诚为您服务




