

Target:Midline-1
Fields:Ubiquitin mediated proteolysis
Gene Name:MID1
Protein Name:Midline-1
Human Gene Id:4281
Human Swiss Prot No:O15344
Mouse Gene Id:17318
Mouse Swiss Prot No:O70583
Rat Gene Id:54252
Rat Swiss Prot No:P82458
Immunogen:The antiserum was produced against synthesized peptide derived from human TRI18. AA range:71-120
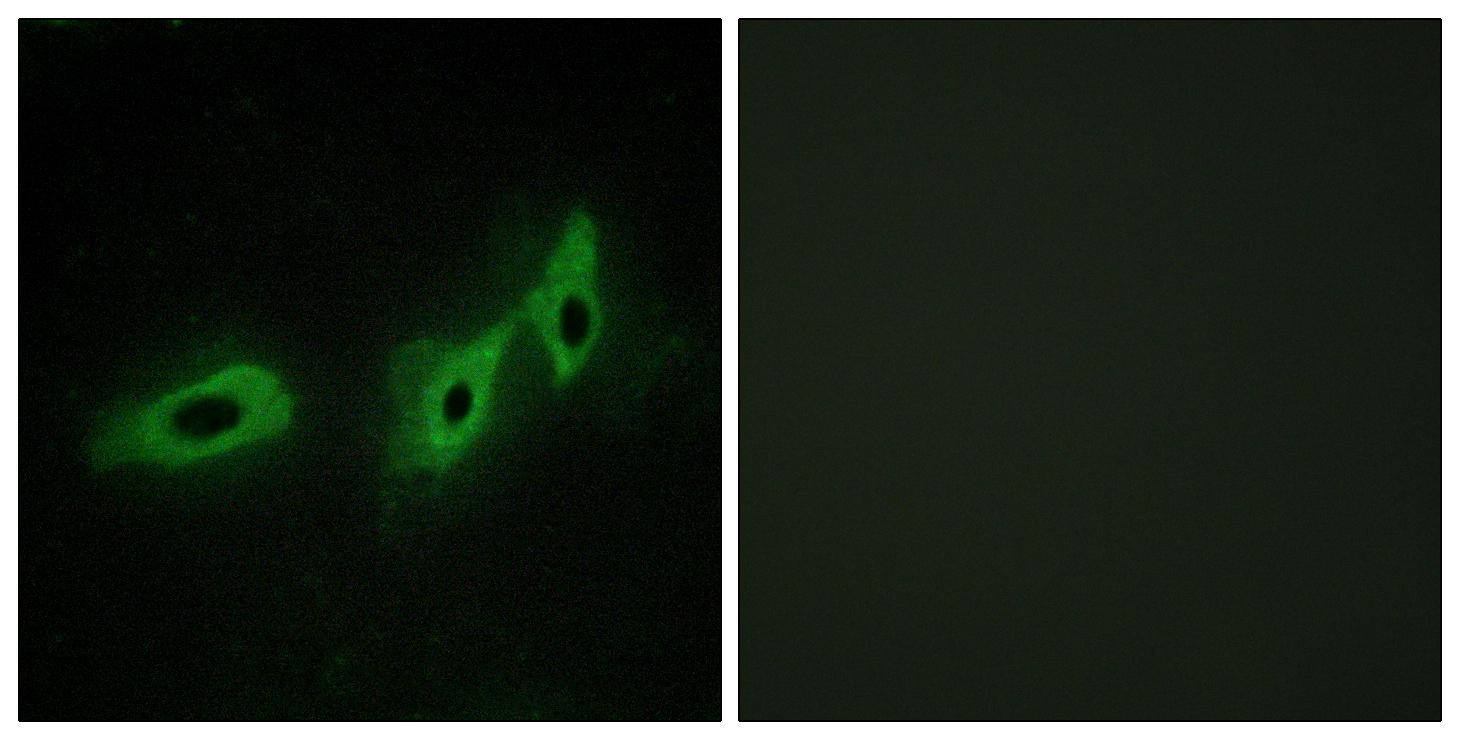
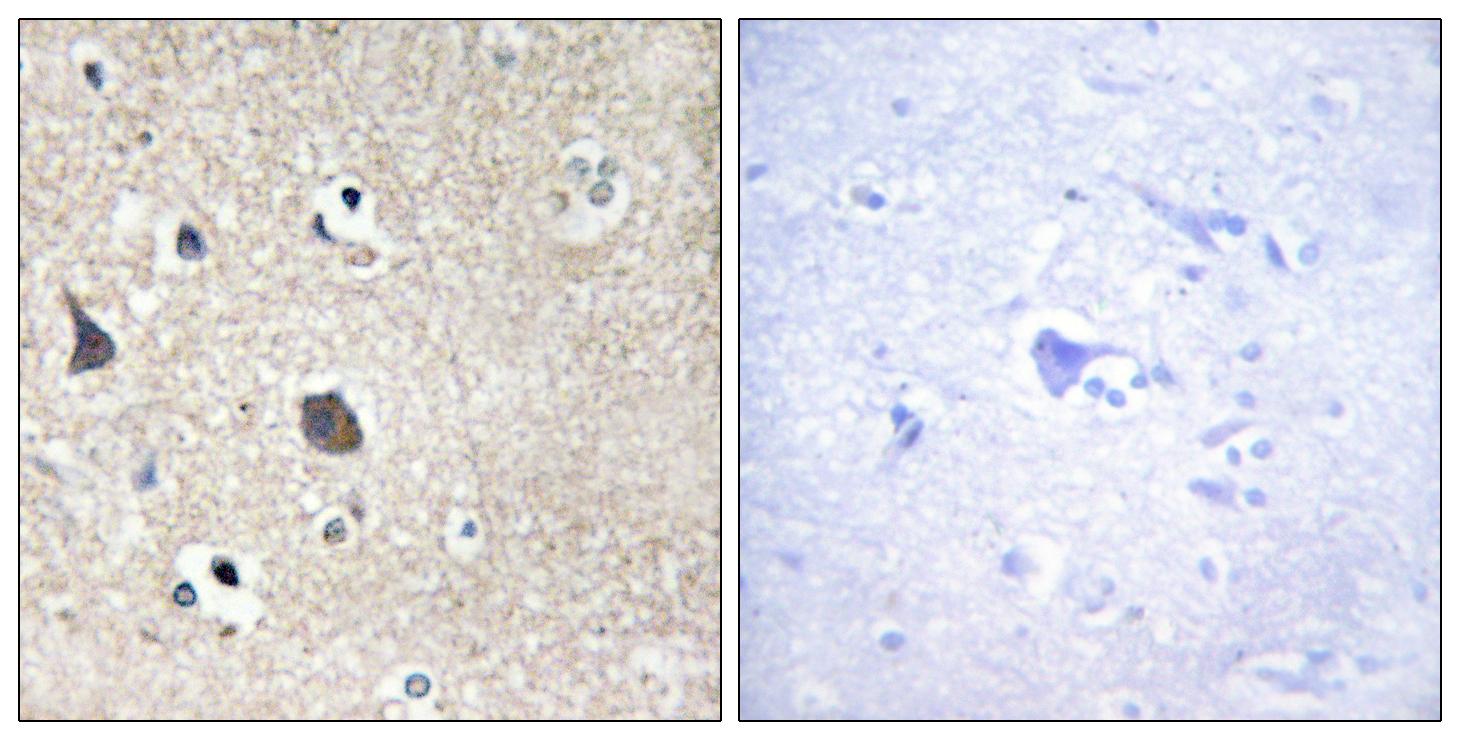
Specificity:Midline-1 Polyclonal Antibody detects endogenous levels of Midline-1 protein.
Formulation:Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Source:Polyclonal, Rabbit,IgG
Dilution:WB 1:500 - 1:2000. IHC 1:100 - 1:300. IF 1:200 - 1:1000. ELISA: 1:40000. Not yet tested in other applications.
Purification:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Concentration:1 mg/ml
Storage Stability:-15°C to -25°C/1 year(Do not lower than -25°C)
Other Name:MID1;FXY;RNF59;TRIM18;XPRF;Midline-1;Midin;Midline 1 RING finger protein;Putative transcription factor XPRF;RING finger protein 59;Tripartite motif-containing protein 18
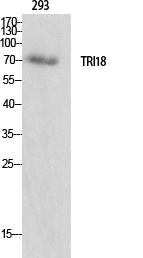
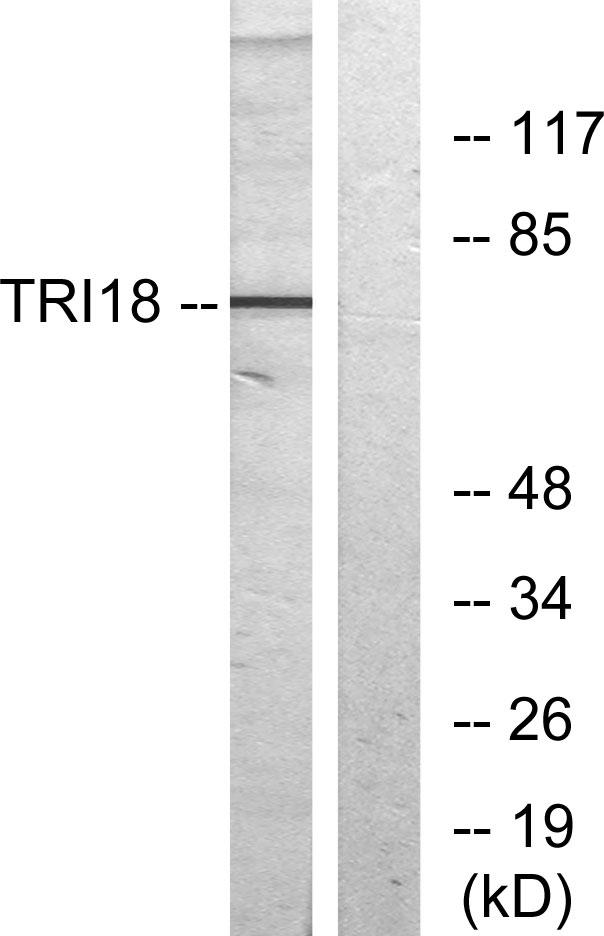
Observed Band(KD):75kD
Background:midline 1(MID1) Homo sapiens The protein encoded by this gene is a member of the tripartite motif (TRIM) family, also known as the 'RING-B box-coiled coil' (RBCC) subgroup of RING finger proteins. The TRIM motif includes three zinc-binding domains, a RING, a B-box type 1 and a B-box type 2, and a coiled-coil region. This protein forms homodimers which associate with microtubules in the cytoplasm. The protein is likely involved in the formation of multiprotein structures acting as anchor points to microtubules. Mutations in this gene have been associated with the X-linked form of Opitz syndrome, which is characterized by midline abnormalities such as cleft lip, laryngeal cleft, heart defects, hypospadias, and agenesis of the corpus callosum. This gene was also the first example of a gene subject to X inactivation in human while escaping it in mouse. Multiple different transcript variants are generated by alternate splicing; however, t
Function:disease:Defects in MID1 are the cause of Opitz syndrome type I (OS-I) [MIM:300000]. OS-I is an X-linked recessive disorder characterized by hypertelorism, genital-urinary defects such as hypospadias in males and splayed labia in females, lip-palate-laryngotracheal clefts, imperforate anus, developmental delay and congenital heart defects. OS-I mutations produce proteins with a decreased affinity for microtubules.,function:May have E3 ubiquitin ligase activity which targets the catalytic subunit of protein phosphatase 2 for degradation.,induction:A retroviral element acts as an alternative tissue-specific promoter for this gene. The LTR of an HERV-E element enhances the expression in placenta and embryonic kidney.,PTM:Phosphorylated on serine and threonine residues.,similarity:Belongs to the TRIM/RBCC family.,similarity:Contains 1 B30.2/SPRY domain.,similarity:Contains 1 COS domain.,simil
Subcellular Location:Cytoplasm . Cytoplasm, cytoskeleton . Cytoplasm, cytoskeleton, spindle . Microtubule-associated. It is associated with microtubules throughout the cell cycle, co-localizing with cytoplasmic fibers in interphase and with the mitotic spindle and midbodies during mitosis and cytokinesis.
Expression:In the fetus, highest expression found in kidney, followed by brain and lung. Expressed at low levels in fetal liver. In the adult, most abundant in heart, placenta and brain.
商品信息已成功复制,启研竭诚为您服务




