Target:APTX
Gene Name:APTX AXA1
Protein Name:Aprataxin (EC 3.-.-.-) (Forkhead-associated domain histidine triad-like protein) (FHA-HIT)
Human Gene Id:54840
Human Swiss Prot No:Q7Z2E3
Mouse Swiss Prot No:Q7TQC5
Rat Swiss Prot No:Q8K4H4
Immunogen:Synthesized peptide derived from human protein . at AA range: 11-60
Specificity:APTX Polyclonal Antibody detects endogenous levels of protein.
Formulation:Liquid in PBS containing 50% glycerol, and 0.02% sodium azide.
Source:Polyclonal, Rabbit,IgG
Dilution:WB 1:500-2000 ELISA 1:5000-20000
Purification:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Concentration:1 mg/ml
Storage Stability:-15°C to -25°C/1 year(Do not lower than -25°C)
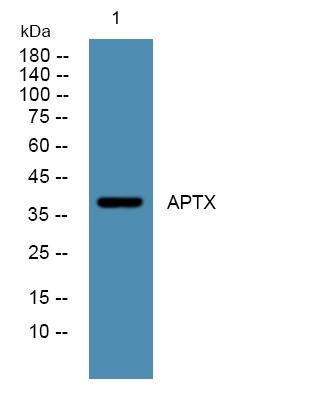
Observed Band(KD):39kD
Background:aprataxin(APTX) Homo sapiens This gene encodes a member of the histidine triad (HIT) superfamily. The encoded protein may play a role in single-stranded DNA repair through its nucleotide-binding activity and its diadenosine polyphosphate hydrolase activity. Mutations in this gene have been associated with ataxia-ocular apraxia. Alternatively spliced transcript variants have been identified for this gene.[provided by RefSeq, Aug 2010],
Function:disease:Defects in APTX are a cause of coenzyme Q10 deficiency [MIM:607426]. Coenzyme Q10 deficiency is an autosomal recessive disorder with variable manifestations. It can be associated with three main clinical phenotypes: a predominantly myopathic form with central nervous system involvement, an infantile encephalomyopathy with renal dysfunction and an ataxic form with cerebellar atrophy. Coenzyme Q10 deficiency due to APTX mutations is typically associated with cerebellar ataxia.,disease:Defects in APTX are the cause of ataxia-oculomotor apraxia syndrome (AOA) [MIM:208920]. AOA is an autosomal recessive syndrome characterized by early-onset cerebellar ataxia, oculomotor apraxia, early areflexia and late peripheral neuropathy.,domain:The C2H2-type zinc finger mediates DNA-binding.,domain:The FHA-like domain mediates interaction with NCL; XRCC1 and XRCC4.,domain:The histidine triad, als
Subcellular Location:Nucleus, nucleoplasm . Nucleus, nucleolus . Upon genotoxic stress, colocalizes with XRCC1 at sites of DNA damage (PubMed:15380105). Colocalizes with MDC1 at sites of DNA double-strand breaks (PubMed:20008512). Interaction with NCL is required for nucleolar localization (PubMed:16777843). .; [Isoform 12]: Cytoplasm .
Expression:Widely expressed; detected in liver, kidney and lymph node (at protein level) (PubMed:14755728). Isoform 1 is highly expressed in the cerebral cortex and cerebellum, compared to isoform 2 (at protein level) (PubMed:14755728). Widely expressed; detected throughout the brain, in liver, kidney, skeletal muscle, fibroblasts, lymphocytes and pancreas (PubMed:15276230, PubMed:11586299, PubMed:11586300).
商品信息已成功复制,启研竭诚为您服务